
Sickle Cell Disease (SCD) is a complex medical entity, associated with significant morbidity and mortality. Patients with SCD suffer from acute and chronic pain, along with a seemingly endless list of additional complications (from hemolysis and sequestration, to aplastic crises and acute chest syndrome). The associated morbidity of SCD is apparent, but these patients also have an alarming decrease in life expectancy, with recent estimates ranging from 28-49 years. Nationally, there is an estimated 5000 patients who suffer from SCD in Canada, and this number is likely to quickly increase given high rates of immigration from countries with high prevalence of SCD, combined with the increased survival associated with improved medical care.
From an Emergency Department (ED) perspective, many providers find the disease and its associated complications challenging and cumbersome. This summary will provide an abbreviated approach to SCD pathophysiology, vaso-occlusive pain crisis (VOC), and acute chest syndrome (AChS) in the ED.
Pathophysiology

Table 1 – Genotypes and phenotypes of possible SCD and SC trait. Taken from Lovett et al.
SCD’s associated pathophysiology is founded in the presence of an abnormally shaped and abnormally functioning HgBS subunit, which when exposed to various physiologic stressors such as hypoxia, causes hemolysis and micro/macrovascular occlusion leading to ischemia, inflammation, and eventual reperfusion injury. Over time, this manifests clinically as hemolytic crises, acute and chronic pain, and a multitude of other associated SCD complications.
As depicted in Table 1, SCD is classically caused by a select few combinations of HgB, including SS disease, SC, as well as HgBS combined with certain subtype of B-thalassemia. Generally, the frequency and severity of clinical manifestations associated with SCD correlate well with the concentration of circulating HgBS. This becomes important when considering patients who receive regularly scheduled exchange transfusions to maintain [HgBS] <30-40% to prevent associated complication of SCD (eg. VOC, stroke, sequestration, etc.)
An important entity to consider in the ED is the sickle cell trait, which results from a combination of HgBS and a normally functioning HgB subunit (eg. HgBA). Traditionally, it has been taught that patients with sickle cell trait are at minimal risk for associated clinical sequelae. However, there are important exceptions to this rule when it comes to ED providers:
- Sickle trait increases risk of rhabdomyolysis and sudden death with prolonged exertion or exercise.
- Sickle trait increases risk of splenic infarcts with prolonged hypoxia or altitude.
- Sickle trait patients are at similar increased risk for glaucoma and increased IOP when presenting with hyphema.3
Vaso-occlusive Crisis
The most common reason SCD patients present to ED is for the evaluation and treatment of acute VOC. When approaching SCD with acute pain, there are a few key overarching themes:
- VOC is a diagnosis of exclusion, and will often be described as the patient’s “usual” pain.
- If there is anything different or abnormal about their pain or associated symptoms, consider other etiologies.
- VOC is classically described as severe, continuous, and progressive pain in the back and/or extremities. It can be precipitated by physiologic stress, illness/injury, or have no clear precipitant whatsoever.
- Once the diagnosis of VOC is made, your goal is to initiate prompt and effective analgesia.
“Drug-Seeking”
Often when SCD patients present in pain, they manifest behaviours and signs which are common amongst drug-seeking patients in the ED, including the absence of tachycardia and/or hypertension.4 Commonly, SCD patients are taught from an early age to employ distraction techniques to cope with severe pain, including talking/texting friends and using their smartphones.
Unfortunately, due to the above reasons, it is a common phenomenon for care providers to stigmatize SCD patients as possible “drug-seekers,” and this has been shown to negatively effect the care they receive as well as their willingness to seek care within the ED.5–7
In reality, multiple quantitative and qualitative studies estimate the proportion of “drug-seeking” in SCD patients at 5%, which is exceedingly low.6 As a result, it is recommended to treat these patients with prompt and aggressive analgesia, and communicate any drug seeking concerns with their primary hematologist.
Are there investigations/bloodwork that can help diagnose or rule out VOC?
The short answer is no. Markers of hemolysis (LDH, haptoglobin, bilirubin, etc.) lack sensitivity: if positive they may increase the likelihood of VOC, but if negative they do not effectively rule out the possibility of VOC.
There is an exception to this rule: the concentration of HgBS. Unfortunately, this test is not readily available for patients presenting to the ED; however, it has important consequences for patients on monthly exchange transfusions:
- If [HgBS] <30-40% (ie. SCD patient receiving monthly exchange transfusions) the chances of a patient experiencing pain associated with VOC is effectively zero.
- Locally, all SCD patients who have demonstrated “drug-seeking” behaviours in the past are on monthly exchange transfusions.
- As such—assuming the patient has not missed this month’s exchange transfusion—when presenting to ED in pain, VOC is effectively ruled out, and their pain should be managed using this specific patient’s acute pain protocol as deligniated by the chronic pain clinic (founded in the avoidance of parenteral opioids).
As an important aside, below is a useful algorithm for the interpretation of HgB and reticulocyte count when evaluating the SCD patient in the ED.
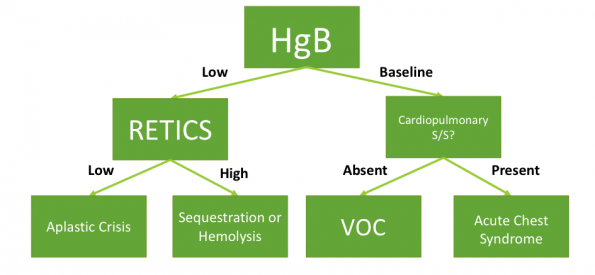
Figure 1 – Adapted from Raam et al., 2016.
Treatment of VOC
Cornerstones:
- Adjuncts:
- Warm blankets or hot water bottles. Local anecdotal evidence and expert opinion suggest that heat improves pain scores.
- Foundational analgesia:
- Guidelines recommend approaching VOC similarly to cancer pain, using the WHO analgesia ladder.9,10
- Prompt Tylenol and NSAID dosing if no other contraindications exists.
- Opioids:
- Guidelines recommend parenteral morphine or hydromorphone.
- Avoid IM dosing given the unreliable pharmacokinetics and pharmacodynamics.
- Avoid Meperidine (Demerol) due to multiple drug-drug interactions and risk of neurotoxic metabolite accumulation in SCD patients.
- Target initiation of parenteral opioids within 30 minutes of triage or 60 minutes of registration.
- Target re-assessment of pain every 15-30 minutes, with consideration of dose escalation by 25%.9
- Approach to initial dosing:
- Local hematology practice is to double patient’s scheduled PO opioid dose at home, then convert to IV/SC.
- IV Fluids:
- There exists some correlation between IV fluids in SCD VOC and congestive atelectasis/pulmonary edema leading to AChS.11
- Bolus IV fluids only if patient is hypovolemic.
- PO hydration is preferred. IV maintenance fluids are only indicated if patient is NPO or unable to tolerate PO hydration.
- O2
- No evidence showing benefit in VOC.
- Case series showing oxygen-related decrease in hematopoiesis requiring transfusions.12
- Guidelines recommend apply O2 if spO2 ≤95%.9,10
Acute Chest Syndrome
Acute chest syndrome (AChS) is an intimidating entity for many ED clinicians. It is the second most common reason for admission and the leading cause of mortality in SCD patients, with case fatality rates ranging from 4 – 25%.9 The majority of information regarding etiology, signs, symptoms, and outcomes is taken from two robust cohort groups: MACSS and CSSCD.13,14
The etiology of adult AChS is predominantly infectious, but often multifactorial, including fat embolism, and pulmonary thrombosis/embolism. As shown in Table 2, there is a large predominance of atypical organisms, and given that the majority of these patients are functionally asplenic, they are also at significant risk for infection with encapsulated bacteria.
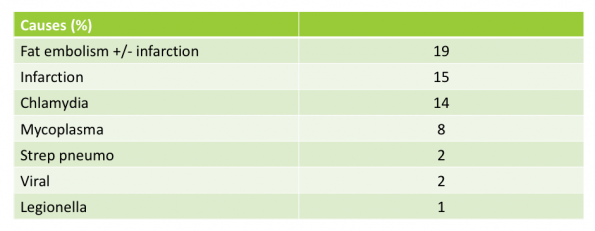
Table 2 – Abbreviated from MACSS data.
When to worry about AChS?
Classically, AChS is defined as:
- Chest pain
AND
- Consolidation on CXR
NIH guidelines recommend considering a workup for AChS when SCD patients present with lower respiratory tract infection symptomatology.
It is important to consider PE when working up these patients given an overall increased incidence in SCD and AChS compared to the general population. If clinically suspected, there are a few caveats to diagnostic approach:
- Classic PE risk stratification tools (Wells, Geneva) are NOT VALIDATED OR INDICATED for these patients.
- In addition to this, SCD alone is highly correlated with having a baseline positive D-dimer. Therefore, D-dimer is unlikely to be helpful and is NOT INDICATED for these patients.
- If you are clinically concerned about PE in a SCD patient, you must consider imaging, such as CT PE.
Treatment of AChS
Cornerstones:
- O2 to maintain spO2 ≥95%
- Antibiotics:
- IV cephalosporin to cover for typicals and encapsulated organisms
- PO macrolide to cover for atypicals
- Admission
- Simple vs. Exchange transfusion
- Overall, this decision should be made in consultation with hematology. There is conflicting evidence regarding benefits and effect on resolution/length of stay.
- Guidelines recommend simple transfusion if: symptomatic AChS with a HgB decrease of >10g/L below baseline, and
- Exchange transfusion if “rapid progression” of AChS (ie. spO2 ≤90% despite supplemental, increasing respiratory effort, or decline in HgB despite simple transfusion)9
Hydroxyurea
Hydroxyurea is, for all intents and purposes, the primary disease-modifying medication for SCD patients. Its primary mechanism of action is to increase the circulating concentration of fetal HgB, and it has a fairly robust body of evidence proving its effectiveness in preventing and/or lessening a multitude of SCD clinical sequelae (summarized in Table 3—taken from the NIH Guidelines)9.
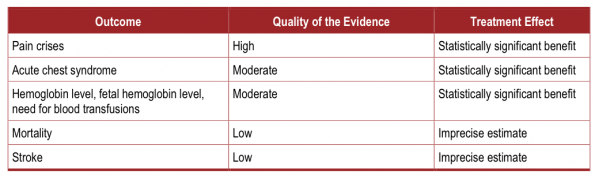
Table 3 – Taken from NIH Sickle Cell Guidelines, 2014.
Currently, hydroxyurea is indicated in the following SCD patients:
- Adults with three or more sickle cell-associated moderate to severe pain crises in a 12-month period. (Strong Recommendation, High-Quality Evidence)
- Adults with sickle-cell associated pain that interferes with daily activities and quality of life. (Strong Recommendation, Moderate-Quality Evidence)
Despite the evidence supporting its use, there remains a phenomenon amongst many SCD patients where many view hydroxyurea as a “bad drug.” Many feel it is not efficacious, and/or carries a multitude of significant side effects.
Clearly emergency providers who are largely unfamiliar with initiating hydroxyurea therapy are not expected to do so. But arguably, the vast majority of SCD patients who present to the ED with VOC should be taking or started on hydroxyurea. This harkens on the importance of the “teachable moment” within the ED. When emergency providers are seeing SCD patients at the peak of their VOC, it is recommended that we discuss the possibility of starting hydroxyurea therapy to decrease VOC frequency and severity. If the patient expresses interest, it is then reasonable to communicate this to the patient’s primary hematologist.
References:
- Ballas, S. K., Pulte, E. D., Lobo, C. & Riddick-Burden, G. Case series of octogenarians with sickle cell disease. Blood 128, 2367–2369 (2016).
- Sickle Cell Disease Association of Canada. (2017). Available at: http://www.sicklecelldisease.ca. (Accessed: 4th November 2017)
- Lovett, P. B., Sule, H. P. & Lopez, B. L. Sickle cell disease in the emergency department. Emerg. Med. Clin. North Am. 32, 629–647 (2014).
- Ernst, A. A., Weiss, S. J., Johnson, W. D. & Takakuwa, K. M. Blood pressure in acute vaso-occlusive crises of sickle cell disease. South. Med. J. 93, 590–592 (2000).
- Labbe, E., Herbert, D. & Haynes, J. ‘Physicians’ Attitude and Practices in Sickle Cell Disease Pain Management’: Erratum. J. Palliat. Care 22, No Pagination Specified (2006).
- Glassberg, J. A. et al. Emergency provider analgesic practices and attitudes toward patients with sickle cell disease. Ann. Emerg. Med. 62, 293–302.e10 (2013).
- Jenerette, C. M., Brewer, C. A. & Ataga, K. I. Care seeking for pain in young adults with sickle cell disease. Pain Manag. Nurs. 15, 324–30 (2014).
- Raam, R., Mallemat, H., Jhun, P. & Herbert, M. Sickle Cell Crisis and You : A How-to Guide. Ann. Emerg. Med. 67, 787–790 (2016).
- National Heart Lung and Blood Institute. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report (EPR). Guidel. Expert Panel Rep. 1–161 (2014). doi:10.1542/peds.2014-2986
- The Canadian Haemoglobinopathy Association. Consensus statement on the care of patients with sickle cell disease in Canada. 1–113 (2015).
- Okomo, U. & Meremikwu, M. M. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane database Syst. Rev. 7, CD005406 (2017).
- Lane, P. K., Embury, S. H. & Toy, P. T. C. Y. Oxygen‐induced marrow red cell hypoplasia leading to transfusion in sickle painful crisis. Am. J. Hematol. 27, 67–68 (1988).
- Vichinsky, B. E. P. et al. Acute Chest Syndrome in Sickle Cell Disease: Clinical Presentation and Course. (2017).
- Vinchinsky, E. Causes and Outcomes of the Acute Chest Synd Ome in Sickle Cell D Is Ease Causes and Outcomes of the Acute Chest Syndrome in Sickle Cell Disease. 1855–1865 (2000). doi:10.1053/j.ajkd.2009.05.012
- Brunson, A., Lei, A., Aaron, S. & White, R. H. Increased incidence of VTE in sickle cell disease patients : risk factors , recurrence and impact on mortality. (2017). doi:10.1111/bjh.14655
- Kabrhel, C. et al. Factors Associated With Positive D-dimer Embolism. 589–597 (2010). doi:10.1111/j.1553-2712.2010.00765.x

